[生信资源与实践】第一次上机-Linux基础命令&RNA_seq
作者:互联网
linux
技巧
- 自动补全:在敲出 文件/目录/命令 的前几个字母之后,按下 tab 键
- 按 上/下 光标键可以在曾经使用过的命令之间来回切换
- 如果想要退出选择,并且不想执行当前选中的命令,可以按 ctrl + c
基础命令
| 命令 | 功能 |
|---|---|
| man command | 查阅 command 命令的使用手册,man 是 manual 的缩写 |
| command --help | 显示 command 命令的帮助信息 |
| ls | 查看当前文件夹下的内容,-lh显示详细信息和文件大小,-a显示隐藏文件 通配符的使用 (*,?,[abc],[a-z]):列出特定文件和文件夹, |
| tree | 命令可以以树状图列出文件目录结构 |
| pwd | 查看当前所在文件夹 |
| cd [目录名] | 切换文件夹 cd ~切换到当前用户的主目录(/home/用户目录) cd .. 切换到上级目录 cd -可以在最近两次工作目录之间来回切换 |
| clear | 清屏 |
| touch [文件名] | 如果文件不存在,新建文件 |
| mkdir [目录名] | 创建目录,-p可以递归创建目录 |
| rm [文件名/ -r 文件夹] | 删除指定的文件名, -r递归地删除目录下的内容,删除文件夹 时必须加此参数 -f强制删除,忽略不存在的文件,无需提示 |
| cp | -i 覆盖文件前提示 -r 若给出的源文件是目录文件,则 cp 将递归复制该目录下的所有子目录和文件,目标文件必须为一个目录名 |
| mv | mv 命令可以用来 移动 文件 或 目录,也可以给 文件或目录重命名 |
| history | 查看历史运行命令 |
| who | 查看当前在线用户 |
| passwd 用户名 | 更改登录密码 |
| ssh 用户名@ip | 远程登录 |
| scp 用户名@ip:文件名或路径 用户名@ip:文件名或路径 | 远程复制文件 |
| which | 可以查看执行命令所在位置 |
| ps aux | process status 查看进程的详细状况 |
| top | 动态显示运行中的进程并且排序 |
| df -h | disk free 显示磁盘剩余空间 |
| du -h [目录名] | disk usage 显示目录下的文件大小 |
查看文件内容
| 命令 | 功能 |
|---|---|
| cat 文件名 | 查看文件内容、创建文件、文件合并、追加文件内容等功能 -b 对非空输出行编号 -n对输出的所有行编号 cat > 1.txt 创建文件覆盖内容 cat >>1.txt 追加内容 |
| more/less 文件名 | 可以用于分屏显示文件内容,每次只显示一页内容 less: 由于more不能后退,就取more的反义词less加上后退功能 |
| grep 搜索文本 文件名 | 模式搜索文本文件内容 -n显示匹配行及行号 -v显示不包含匹配文本的所有行(相当于求反) -i忽略大小写 -c ‘文字’ 文件 →某个字符串出现的个数, 模式查找: ^a行首,搜寻以 a 开头的行 ke$行尾,搜寻以 ke 结束的行 |
| head -n | 显示文件前多少行内容 |
| tail -n | 显示文件末尾多少行内容 |
| cut | cut 命令从文件的每一行剪切字节、字符和字段并将这些字节、字符和字段写至标准输出 -d ‘ ’:自定义分隔符,默认为制表符。 -f :提取哪些字段 cut -f 1,4-7 1.txt 显示1.txt第1列和4到7列内容 |
| sort | 用于将文本文件内容加以排序 sort -k2 -nr test.bed : -k2,优先按第二列排序 -t ‘’:设定间隔符 nr :-n按数字大小排序,默认按照ASCII码排序,-r倒序 -u去掉重复行 |
RNA-seq
1.准备工作
# 将pipelinesh复制到自己的目录下
cp /project/hts-demo/pipeline.sh ./
# 运行pipeline.sh
./pipeline.sh
运行代码
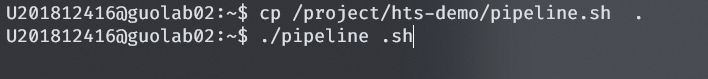
得到demo文件夹
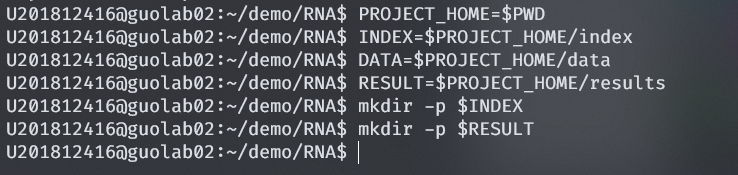
再开一个窗口(方便之后的复制),查看demo/RNA/work.sh文件
2. 变量命名
PROJECT_HOME=$PWD
INDEX=$PROJECT_HOME/index
DATA=$PROJECT_HOME/data
RESULT=$PROJECT_HOME/results
mkdir -p $INDEX
mkdir -p $RESULT
输入命令,对路径进行命名,是为了简化接下来的代码
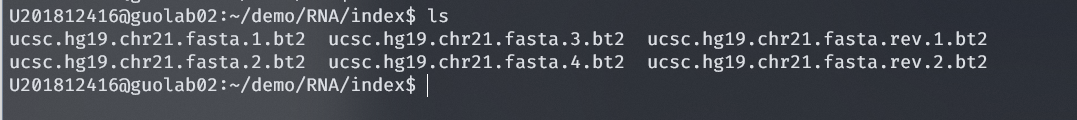
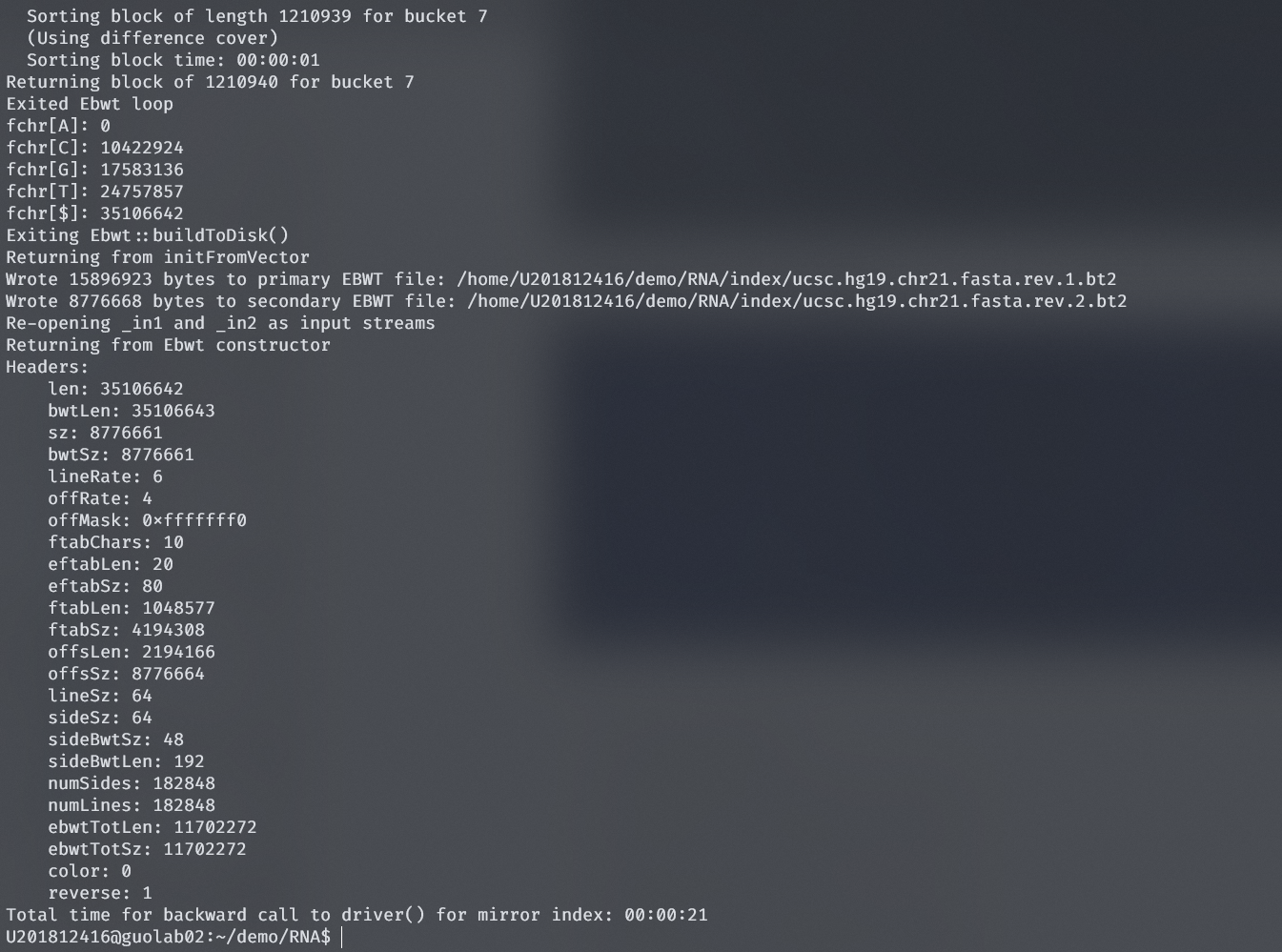
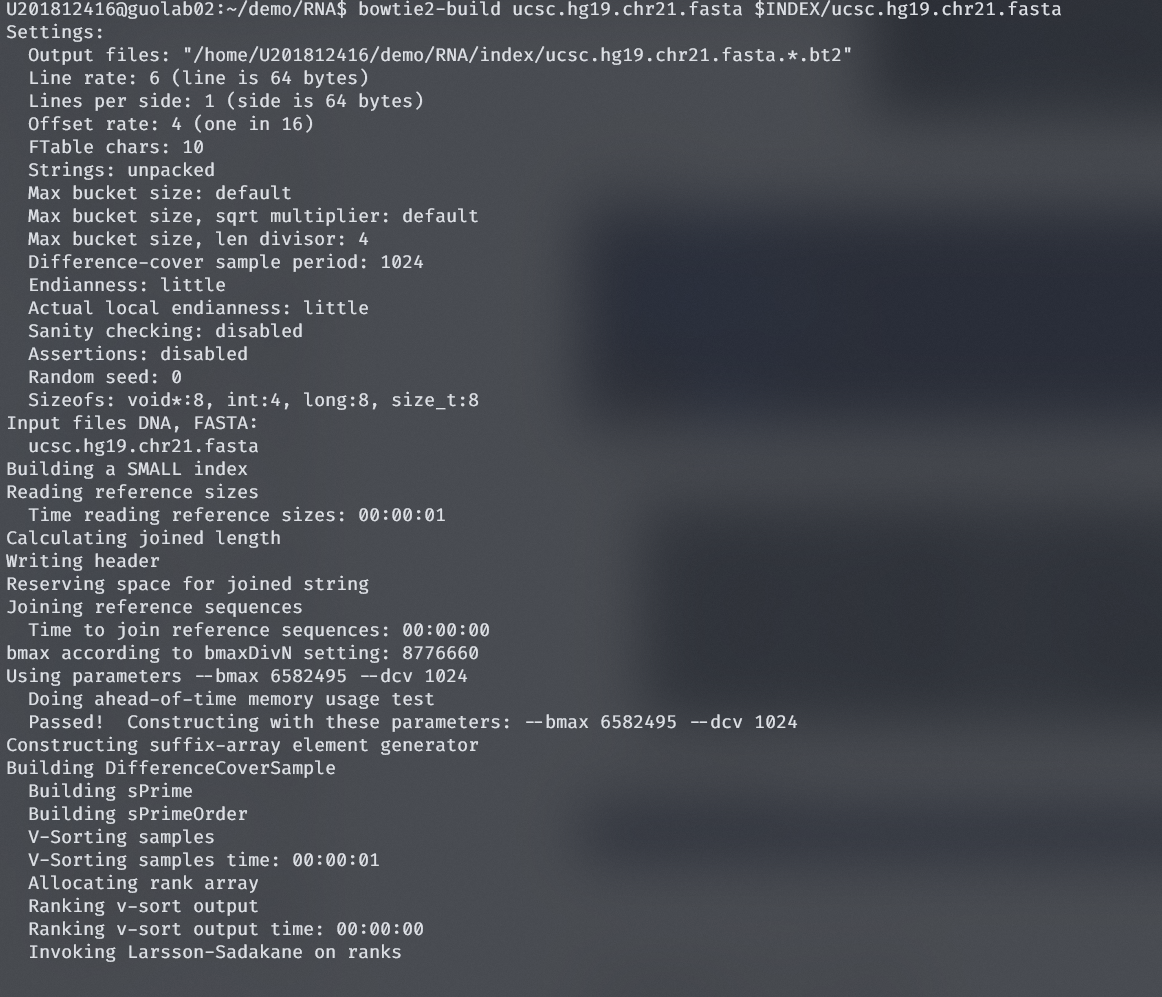
3. 使用Bowtie构建索引
# build bowtie2 index
bowtie2-build ucsc.hg19.chr21.fasta $INDEX/ucsc.hg19.chr21.fasta
运行命令
运行结束
发现index文件多了以bt2结尾的文件
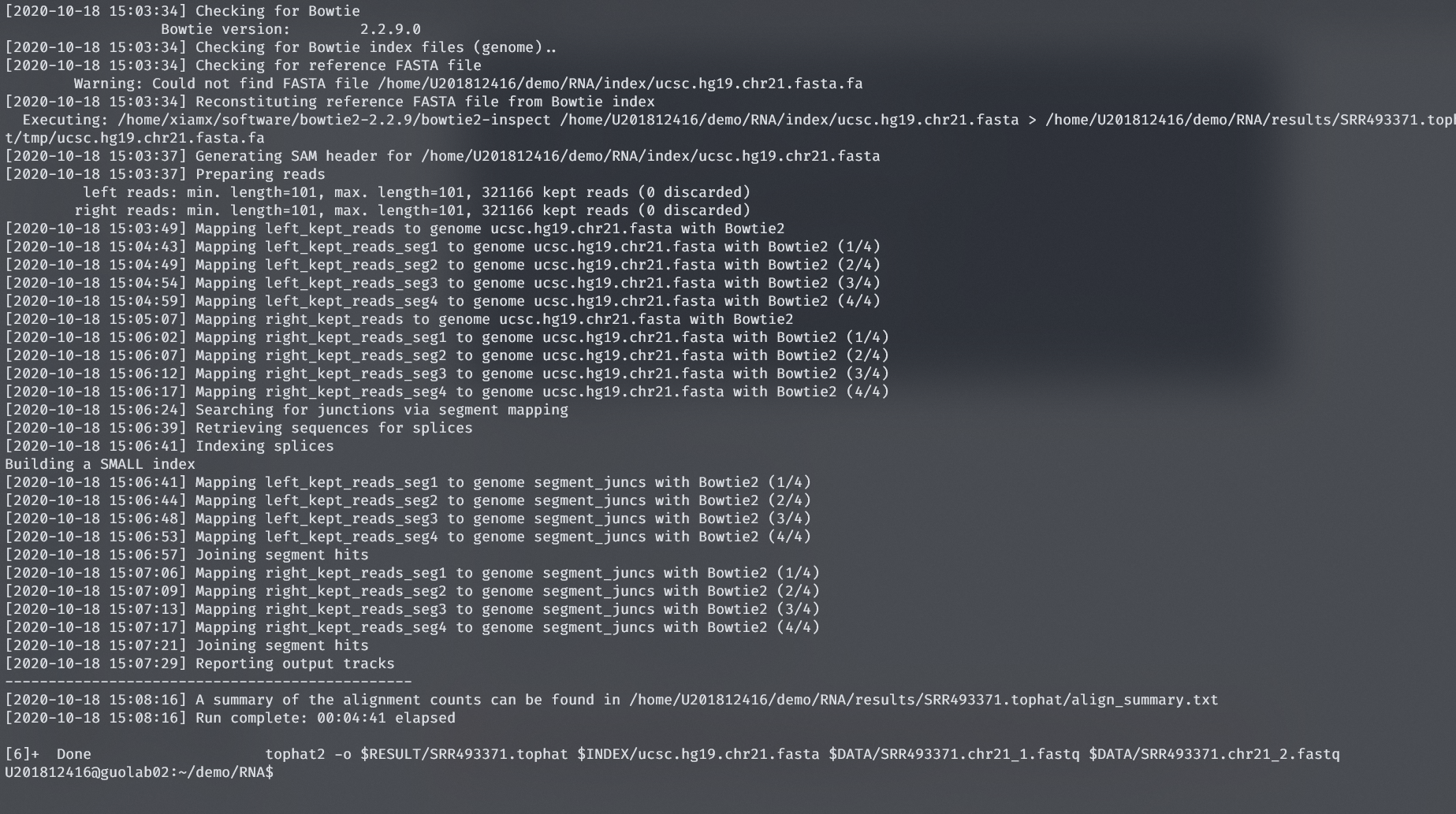
4. 使用tophat将reads片段比对到基因组上
# mapping with tophat2
tophat2 -o $RESULT/SRR493366.tophat $INDEX/ucsc.hg19.chr21.fasta $DATA/SRR493366.chr21_1.fastq $DATA/SRR493366.chr21_2.fastq &
tophat2 -o $RESULT/SRR493367.tophat $INDEX/ucsc.hg19.chr21.fasta $DATA/SRR493367.chr21_1.fastq $DATA/SRR493367.chr21_2.fastq &
tophat2 -o $RESULT/SRR493368.tophat $INDEX/ucsc.hg19.chr21.fasta $DATA/SRR493368.chr21_1.fastq $DATA/SRR493368.chr21_2.fastq &
tophat2 -o $RESULT/SRR493369.tophat $INDEX/ucsc.hg19.chr21.fasta $DATA/SRR493369.chr21_1.fastq $DATA/SRR493369.chr21_2.fastq &
tophat2 -o $RESULT/SRR493370.tophat $INDEX/ucsc.hg19.chr21.fasta $DATA/SRR493370.chr21_1.fastq $DATA/SRR493370.chr21_2.fastq &
tophat2 -o $RESULT/SRR493371.tophat $INDEX/ucsc.hg19.chr21.fasta $DATA/SRR493371.chr21_1.fastq $DATA/SRR493371.chr21_2.fastq &
运行命令
运行结束
比对后得到的6个bam文件:
results/SRR493366.tophat/accepted_hits.bam
results/SRR493367.tophat/accepted_hits.bam
results/SRR493368.tophat/accepted_hits.bam
results/SRR493369.tophat/accepted_hits.bam
results/SRR493370.tophat/accepted_hits.bam
results/SRR493371.tophat/accepted_hits.bam
当我们测序得到的fastq数据map到基因组之后,会得到一个以sam或bam为扩展名的文件。这里,SAM的全称是sequence alignment/map format。而BAM就是SAM的二进制文件,也就是压缩格式的sam文件。
使用命令 samtools view accepted_hits.bam查看SRR493366.tophat/accepted_hits.bam
使用IGV查看
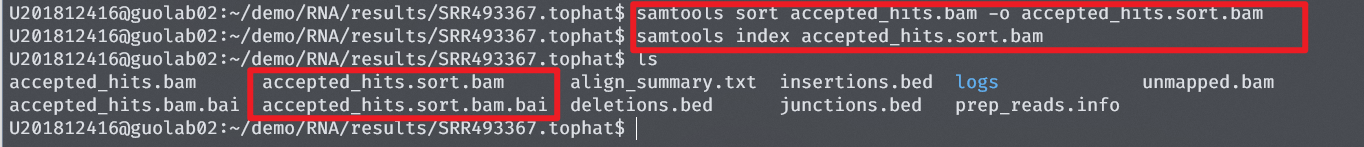
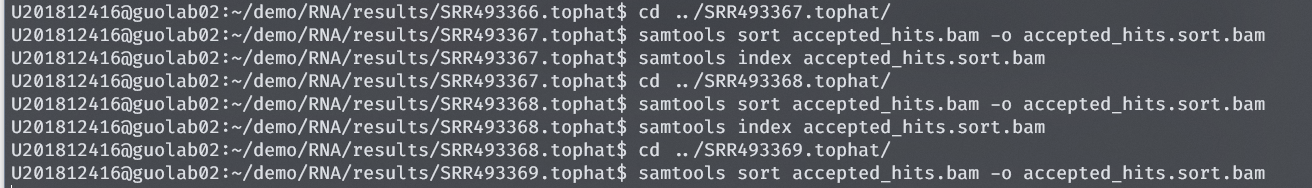
①先使用samtools对6个bam文件进行排序和构建索引
#samtools 对bam进行排序
samtools sort accepted_hits.bam -o accepted_hits.sort.bam
#samtools 对BAM文件建立索引
samtools index accepted_hits.sort.bam
以SRR493366.tophat为例,如下图
然后对其他五个文件夹也是如此处理
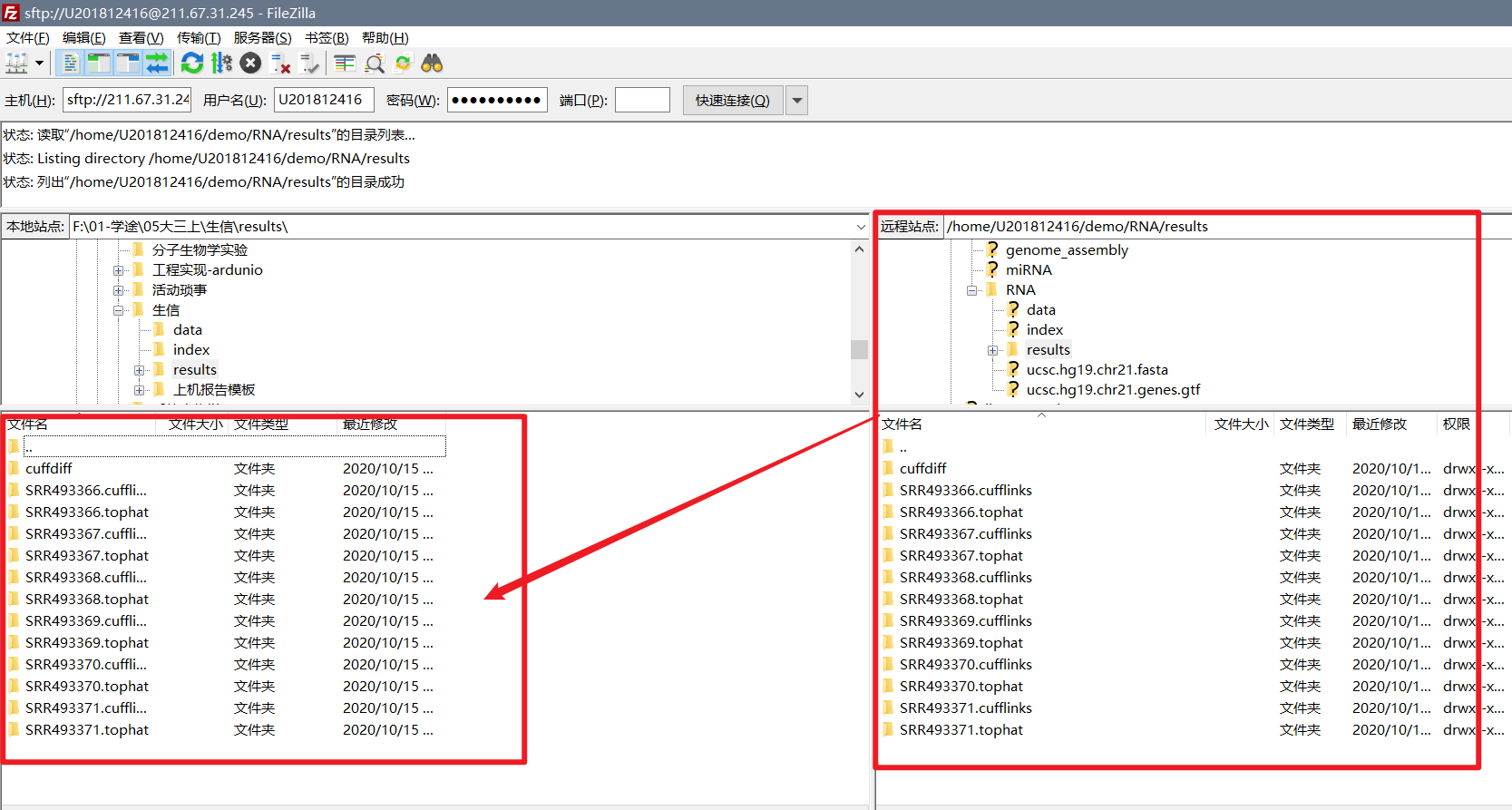
②使用具有sftp功能的软件FileZilla将Linux服务器上的文件传到自己电脑
FileZilla win 32位便携版 [下载地址]
输入Linux服务器ip地址211.67.31.245,自己的用户名,密码,端口22,点击链接即可
 ③安装并打开IGV
③安装并打开IGV
IGV -Win版 included java [ 下载地址]
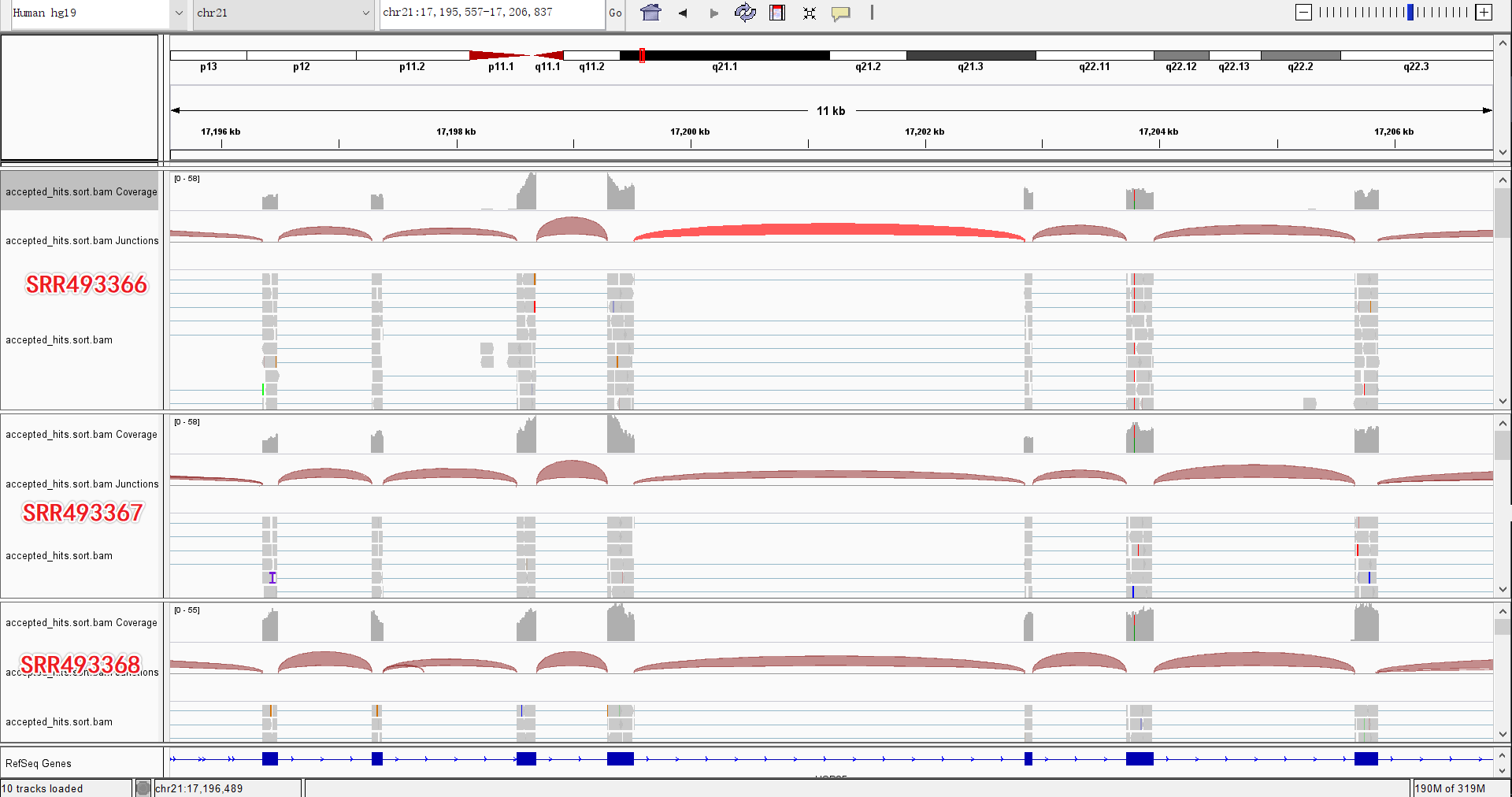
将bam文件导入,选择hg19,chr21,通过调节右上角的页面缩放和点击上方的染色体位置来显示比对区域
④查看比对结果
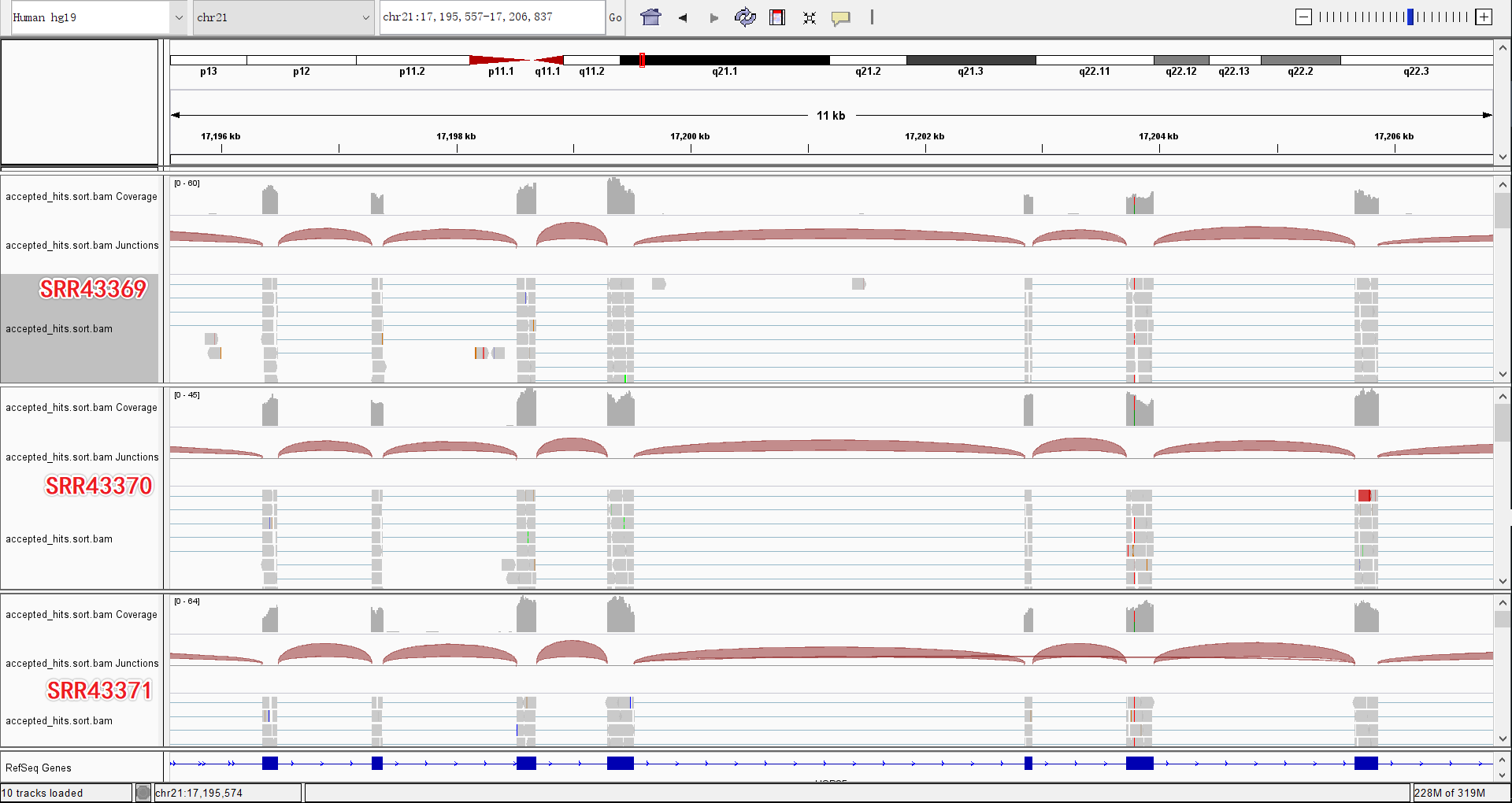
SRR493366、SRR493367、SRR493368的比对结果:(位置-chr21:17,195,557-17,206,837)
SRR493369、SRR493370、SRR4933671的比对结果:(位置-chr21:17,195,557-17,206,837)
载入bam文件后会产生3个相关的tracks:
- Alignment track :显示每个的reads的比对情况
- Coverage track:显示覆盖率和测序深度,如果某核苷酸与参考序列不同(超过20%reads)时,IGV会标出不同的颜色。即:A→绿色;C→蓝色;G→橙色;T→红色。
- Splice Junction Track:提供一个可选的横跨剪切位点(spanning splice junctions)的reads视图。
5. 使用Cufflinks计算表达水平
# calculate expression level
cufflinks -G ucsc.hg19.chr21.genes.gtf -o $RESULT/SRR493366.cufflinks $RESULT/SRR493366.tophat/accepted_hits.bam &
cufflinks -G ucsc.hg19.chr21.genes.gtf -o $RESULT/SRR493367.cufflinks $RESULT/SRR493367.tophat/accepted_hits.bam &
cufflinks -G ucsc.hg19.chr21.genes.gtf -o $RESULT/SRR493368.cufflinks $RESULT/SRR493368.tophat/accepted_hits.bam &
cufflinks -G ucsc.hg19.chr21.genes.gtf -o $RESULT/SRR493369.cufflinks $RESULT/SRR493369.tophat/accepted_hits.bam &
cufflinks -G ucsc.hg19.chr21.genes.gtf -o $RESULT/SRR493370.cufflinks $RESULT/SRR493370.tophat/accepted_hits.bam &
cufflinks -G ucsc.hg19.chr21.genes.gtf -o $RESULT/SRR493371.cufflinks $RESULT/SRR493371.tophat/accepted_hits.bam &
运行命令
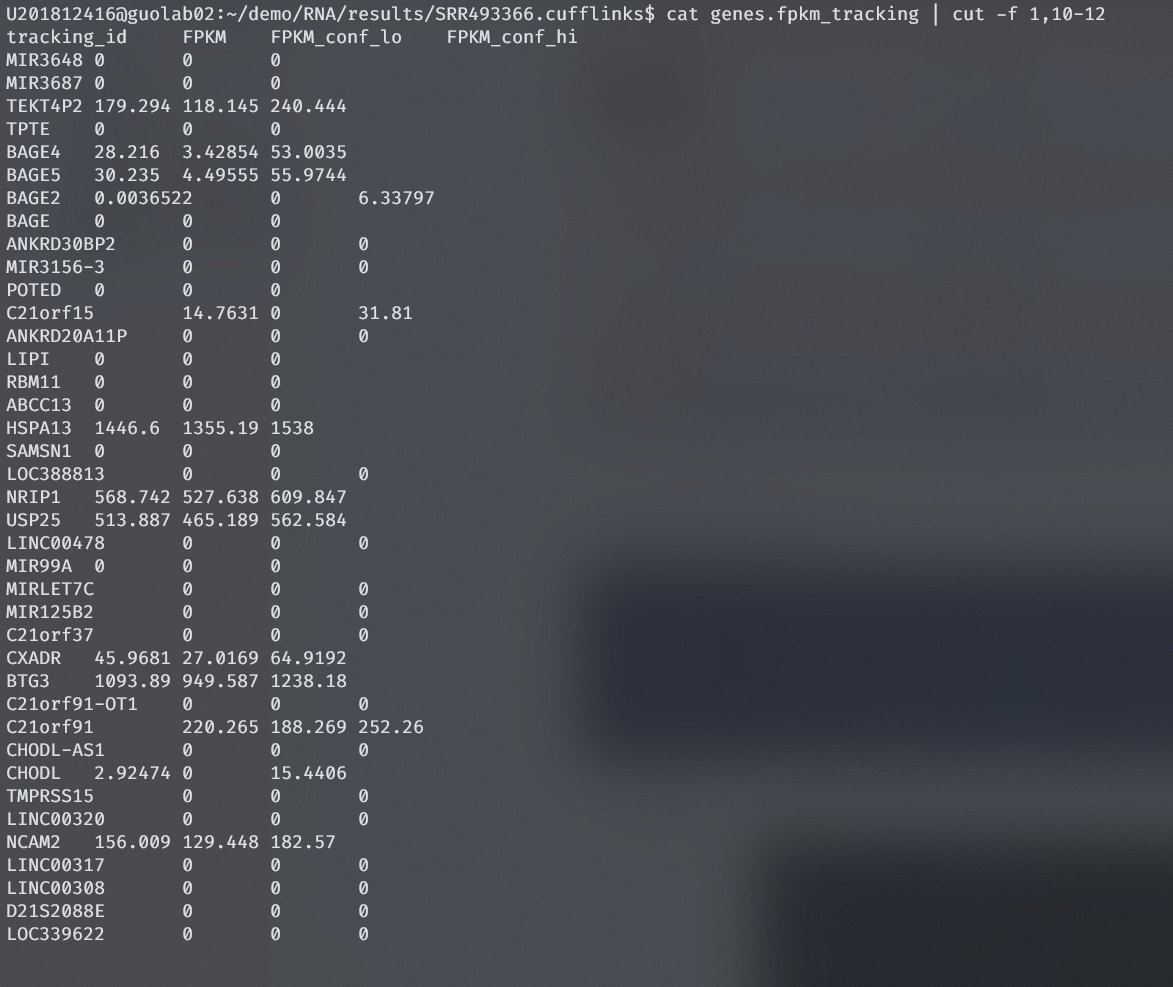
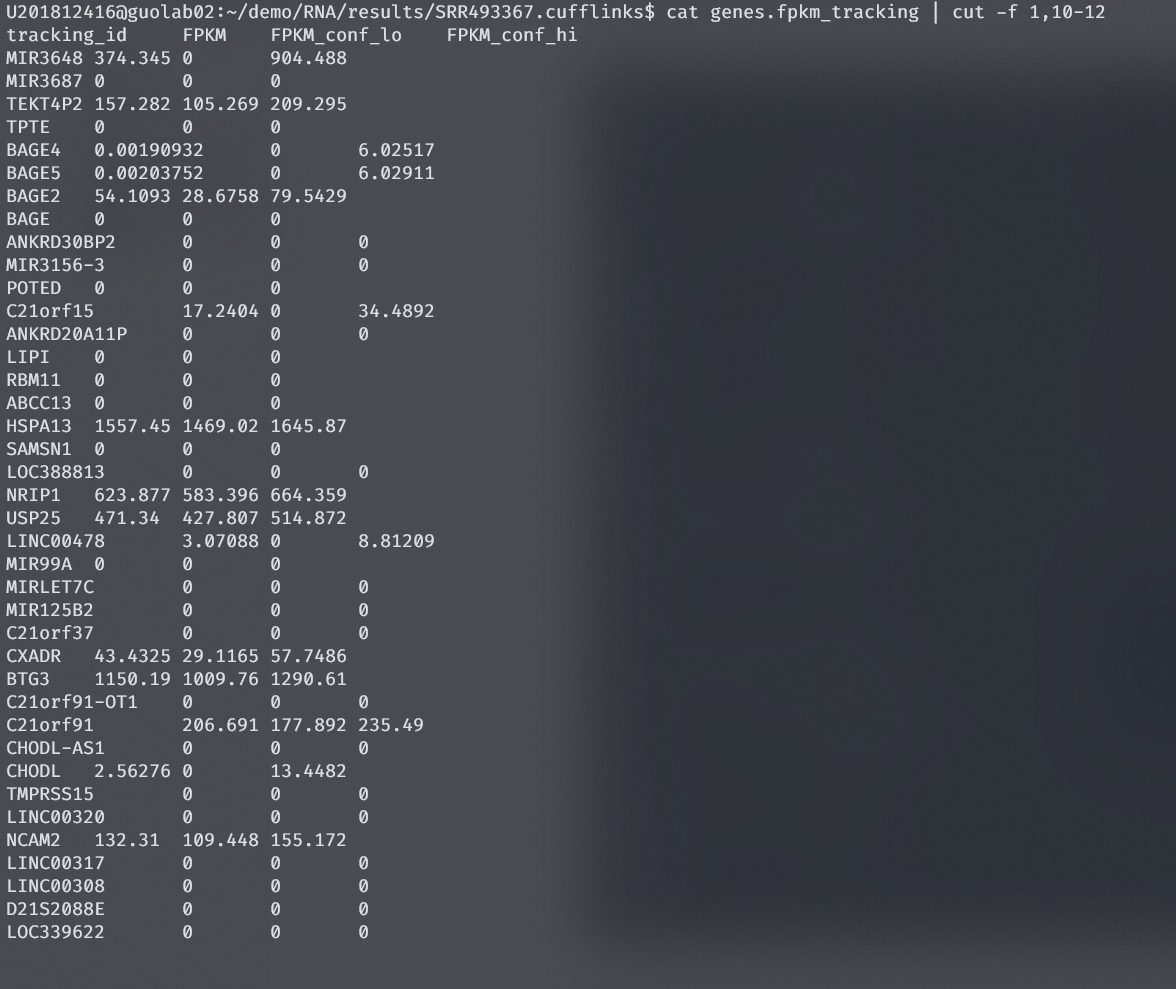
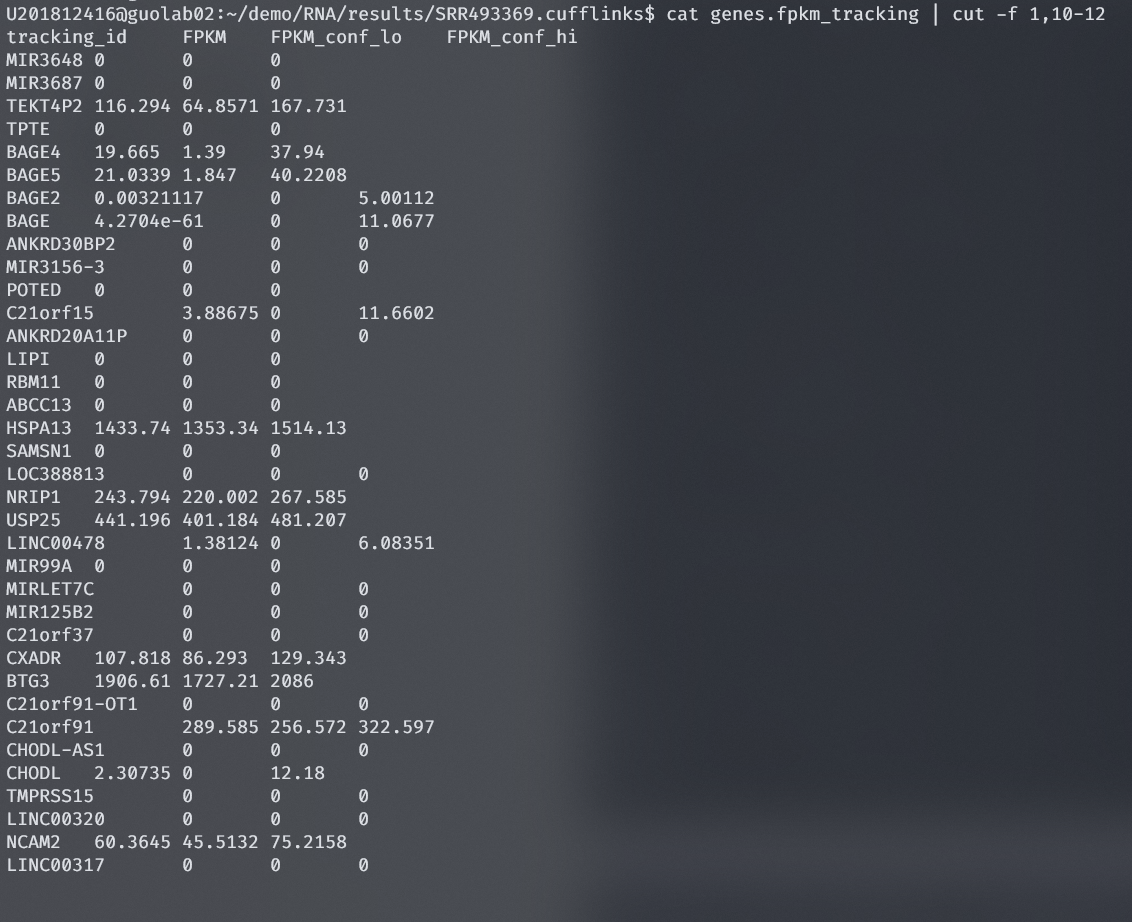
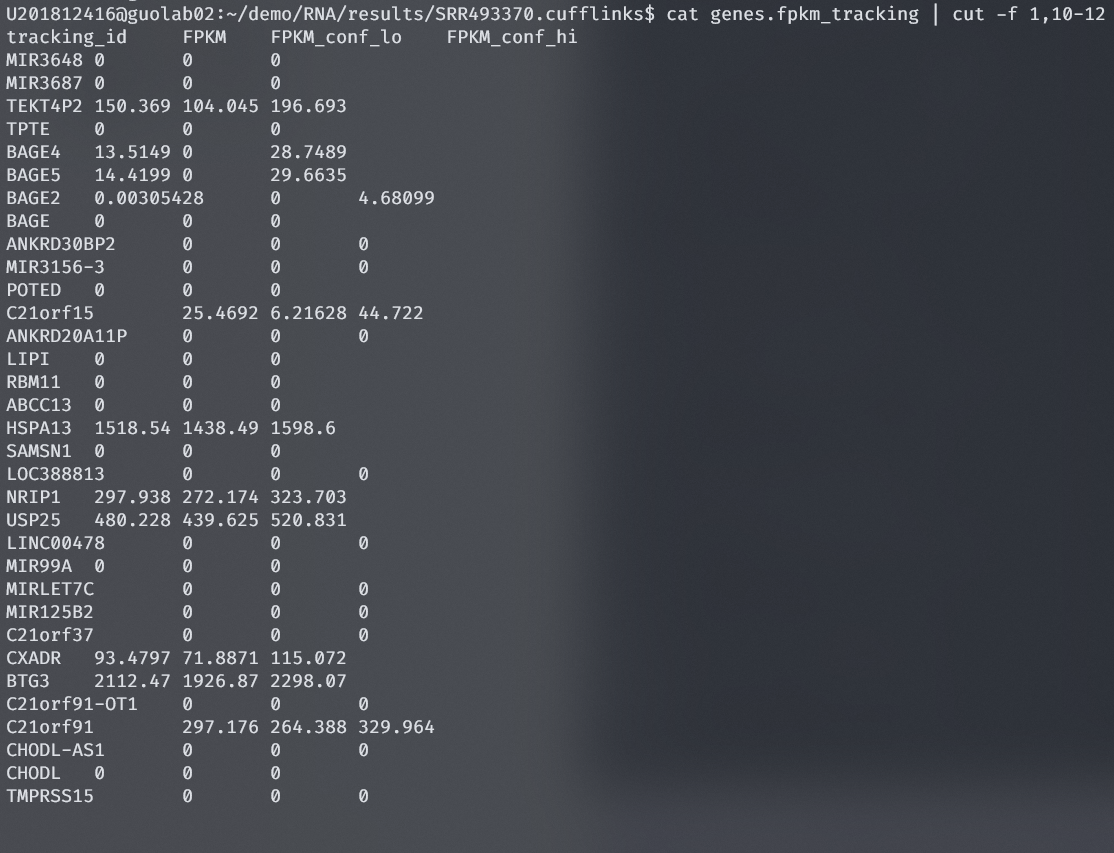
查看结果,使用cat genes.fpkm_tracking | cut -f 1,10-12截取第1列和10-12列查看FPKM值
RPKM = Reads Per Kilobase per Million mapped reads
FPKM = Fragments Per Kilobase per Million mapped reads
RPKM中的R指的是Reads针对单端测序,FPKM中的F是指Fragments, 针对双端测序我们现在测序一般来说都是测双端测序(paired-end sequencing),那么在mapping回参考基因组的时候就会有两条reads,分别是read1和read2,分别来源于建库打断的5' 端和3'端。那么这2条reads就可以在参考基因组上确定1个小的片段,这个片段就叫fragment.
FPKM能够矫正掉gene长度以及测序深度对gene表达定量的影响
可以看到不是所有的基因都表达,不同的样本不同的基因表达水平也不同
results/SRR493366.cufflinks/genes.fpkm_tracking
results/SRR493367.cufflinks/genes.fpkm_tracking
results/SRR493368.cufflinks/genes.fpkm_tracking
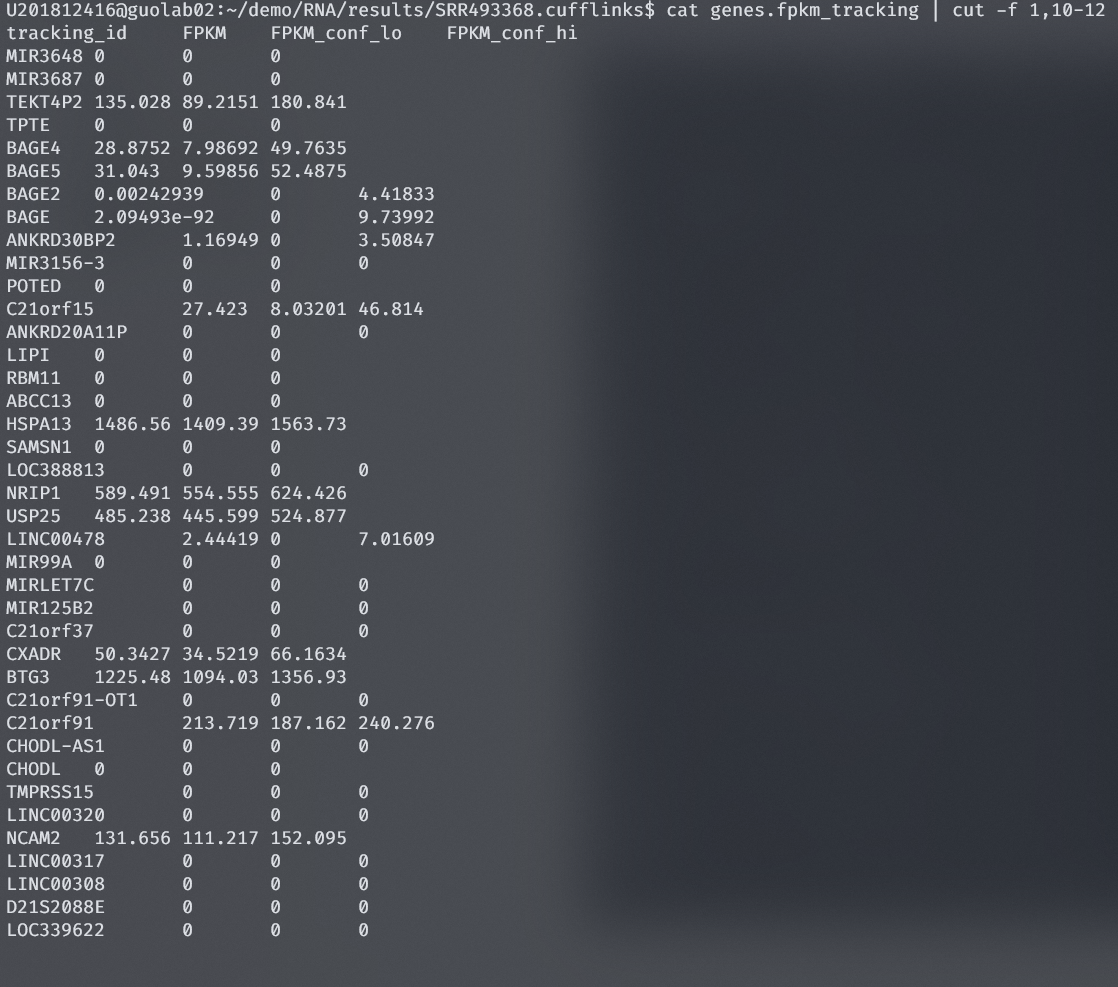
results/SRR493369.cufflinks/genes.fpkm_tracking
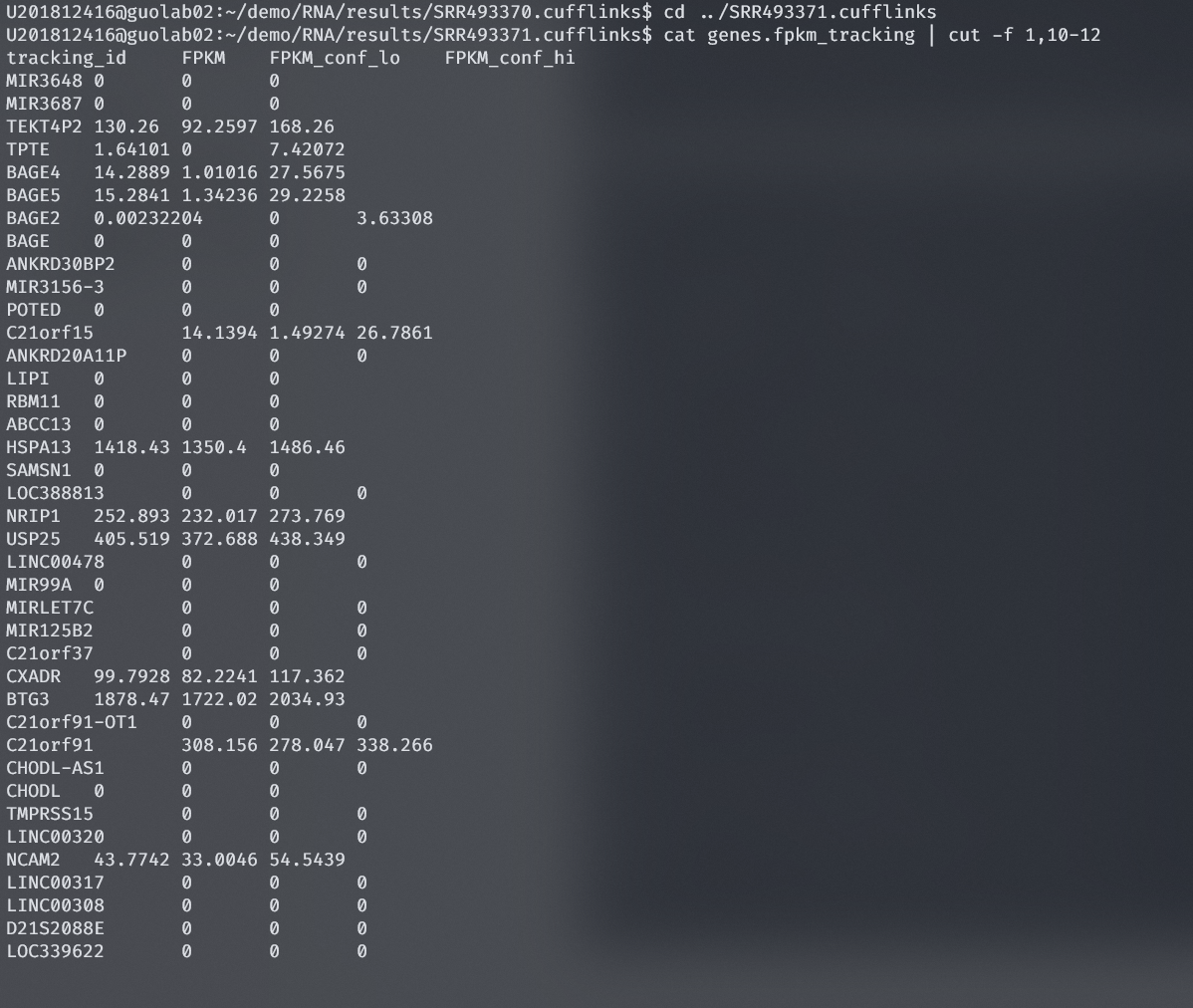
results/SRR493370.cufflinks/genes.fpkm_tracking
results/SRR493371.cufflinks/genes.fpkm_tracking
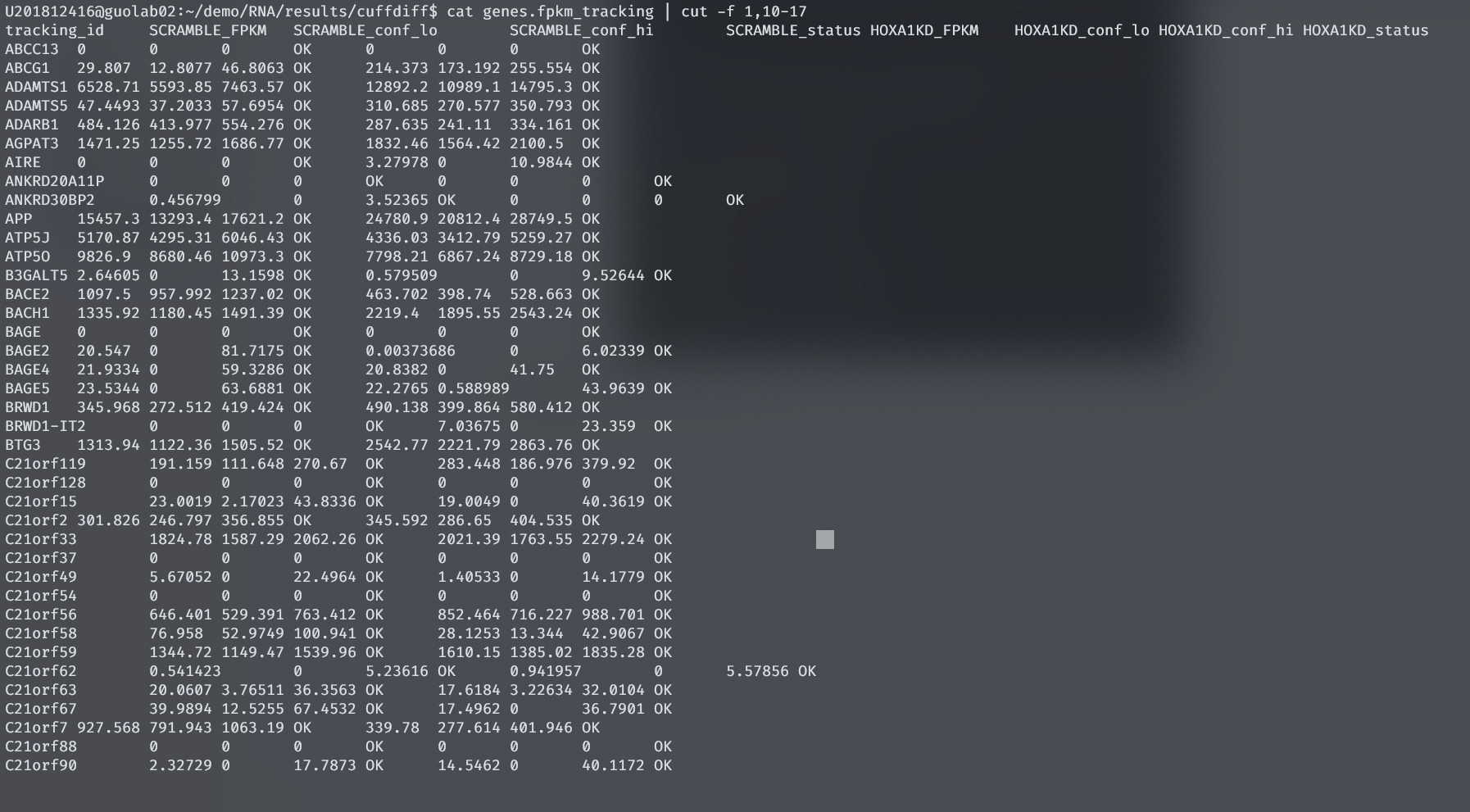
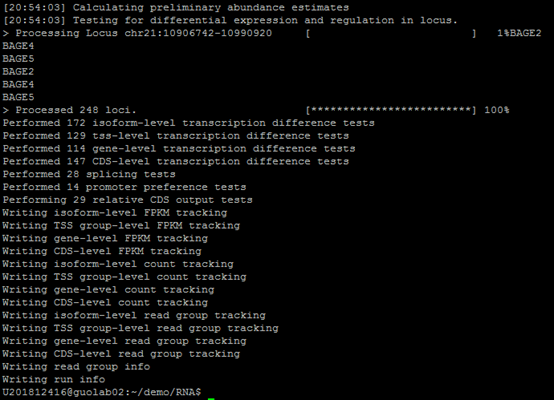
6.使用Cuffdiff—根据比对结果,计算差异表达
# find differentially expressed genes
cuffdiff ucsc.hg19.chr21.genes.gtf \
-o $RESULT/cuffdiff \
-L SCRAMBLE,HOXA1KD \
$RESULT/SRR493366.tophat/accepted_hits.bam,$RESULT/SRR493367.tophat/accepted_hits.bam,$RESULT/SRR493368.tophat/accepted_hits.bam \
$RESULT/SRR493369.tophat/accepted_hits.bam,$RESULT/SRR493370.tophat/accepted_hits.bam,$RESULT/SRR493371.tophat/accepted_hits.bam
运行命令
结果路径results/cuffdiff/genes.fpkm_tracking, 使用cat genes.fpkm_tracking | cut -f 1,10-17查看
标签:hits,accepted,seq,RNA,RESULT,chr21,tophat,bam,生信 来源: https://www.cnblogs.com/achuan-2/p/13861685.html