HiFi全基因组测序技术与实例|HiFi基因组组装软件推荐
作者:互联网
HIFI技术的简介
HiFi reads(High fidelity reads)
- 是Sequel II 三代测序平台推出的兼顾长读长和高准确度的测序序列,一般采用CCS(Circular Consensus Sequencing)模式测序。在这种测序模式下,酶读长一般大于插入片段长度,因此酶会绕着模板进行滚环测序,插入片段会被多次测序。单次测序中造成的随机测序错误,可以通过算法进行自我纠错校正,最终得到高准确度的HiFi reads。
- 要在单次测序中得到更多的HiFi reads往往需要平衡测序的酶读长和插入片段的长度,插入片段太长会导致酶无法进行滚环测序,插入片段太短又牺牲了三代长读长测序的优势。因此HiFi模式测序对酶试剂和建库过程的均一性要求较高。
更多相关内容可以点击关注我的网站
HiFi建库流程
PacBio SMRT测序原理
- 聚合酶捕获文库DNA序列,锚定在零模波导孔底部
- 4种不同荧光标记的dNTP随机进入零模波导孔底部
- 荧光dNTP被激光照射,发出荧光,检测荧光
- 荧光dNTP与DNA模板的碱基匹配,在酶的作用下合成一个碱基
- 统计荧光信号存在时间长短,区分匹配碱基与游离碱基,获得DNA序列
- 酶反应过程中,一方面使链延伸,另一方面使dNTP上的荧光基团脱落
- 聚合反应持续进行,测序同时持续进行
以下为图示:


SMRT测序的两种模式
SMRT测序目前有CLR模式与CCS模式,从图片可以看出CLR是提高了序列长度但是没有兼顾准确度,而CCS模式的话既兼顾了长读长又有很好的准确度,我们通常将准确度大于99%(Q20)的read称为HiFi read
[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-vJrQw3qj-1612082508678)(https://s3.ax1x.com/2021/01/18/scSACF.png)]
下面是CCS模式的具体流程,简单口述一下:
将一对高质量的DNA序列两端加上接头,之后在加入引物以及DNA聚合酶,在此模式下可进行滚环测序,即一对DNA序列被重复测序了多次之后去掉接头和多余的错误得到subreads,最后经过纠错得到HiFi READ
[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-Qa2c3KRt-1612082508680)(https://s3.ax1x.com/2021/01/18/s6zxjs.png)]
HiFI组装实例
首先我们可以看一下HIFI的应用,可以看出HIFI测序技术的出现,使得一些复杂有难度的植物基因组也可以通过测序得出较好的结果,下面主要针对最近的这几篇文章的思路入手,了解一下用与HiFi READ基因组组装的一些软件以及组装的大致流程
水稻、果蝇和人类的基因组组装
下面的话是使用HIFI READS 生成连续,完整和准确的 DE NOVO 组装,分别是水稻、果蝇和人类的基因组组装结果展示,可以看到准确度达到了Q50
六倍体加州红杉基因组
PacBio的科学家利用HiFi测序在两周内完成了基因组高达27Gb的六倍体加州红杉基因组的组装,与以前使用其他技术的针叶树组装相比,使用 PacBio HiFi de novo 组装的加州红杉(Sequoia sempervirens)在3个C (Contiguity, Completeness, and Correctness) 的基因组组装质量方面实现了显着的性能改善。
第一列为PacBio HiFi组装结果只用了6天,相比而言第二列的ONT+short reads组装(也就是目前大多采用的三代+二代组装的策略)花费的时间减少了很多!由于加州红杉为裸子植物而BUSCO评价完整度的参考大多为被子植物所以这个评价的结果不算高也正常。
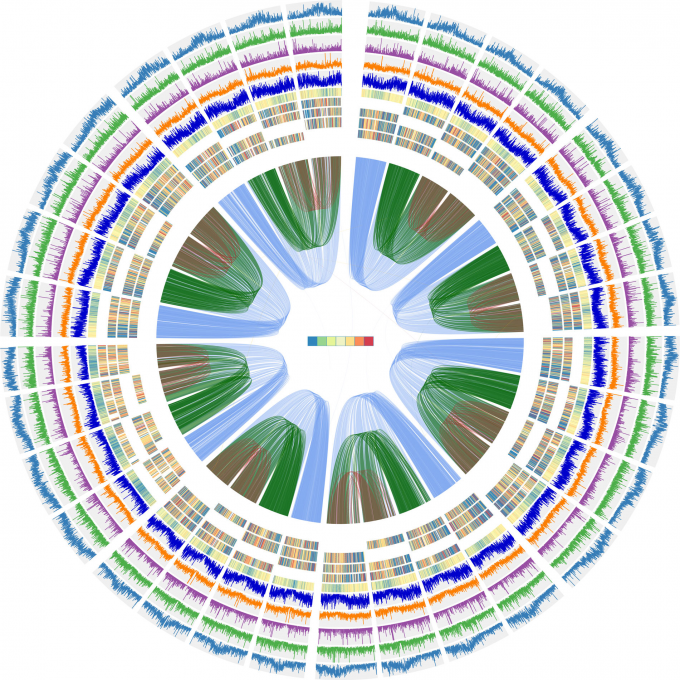
紫花苜蓿基因组组装
Published: 19 May 2020
(1)使用Canu默认参数,利用CCS clean reads组装contigs。组装得到的Contig N50值为459kb,总长度为3.15GB。
(2)使用HiC-Pro将Hi-C reads与contigs 进行比对,产生比对BAM文件。
(3)使用注释的蒺藜苜蓿蛋白作为参考,完全基于同源的策略注释contigs。对138,729个同源基因进行了结构注释。用MCscan用于鉴定contigs和参考基因组之间的共线性。显示紫花苜蓿和蒺藜苜蓿之间的高共线性。
(4)使用内部脚本处理BAM文件,去除等位基因contigs之间的links。使用ALLHiC软件,提取、聚类和重排Contigs (Contigs syntenic与蒺藜苜蓿染色体一致),得到原始的scaffolds。
(5)Juicebox用于以图形和交互方式微调组装的scaffolds。剪裁了40个总长度达1800Mb的scaffolds 。
(6) 基于组装的scaffold,通过Hi-C数据,每个unplaced contig被分配到互作最强的那些contig cluster里。
(7)使用ALLHiC对那些contig clusters再次进行重排和构建scaffold。
(8)使用Juicebox对scaffolds进行微调,并从scaffolds上去除不一致的contigs,产生最终的染色体基因组,其包含32条染色体(8个同源组,每个组中有4个等位基因染色体),总长度为2738Mb,和419Mb未挂载到染色体水平的序列。

全基因组测序的组装流程
pb-assembly:内容包含各个falcon的流程,支持CLR/HiFi的组装,适合各种基因组的组装。
pbgcpp:支持CLR组装的纠错,纠错的raw reads必须是subreads.bam
第三方软件:
HiFi large genome:主要有HiCanu, Hifiasm, 以及Peregrine
CLR large genome:主要有Canu, wtdbg2
Small genome:主要有Flye, Canu
下面主要介绍一下HiFi基因组组装目前推荐的软件:
Hicanu
Genome Research ( IF 11.093 ) Pub Date : 2020-09-01
- PacBio HiFi Reads结合HiCanu能够生成迄今为止最准确,最完整的人类基因组装配体,对基因组连续性、准确性、单倍型检测以及应对复杂片段重复序列均有提升
- HiCanu(Canu专门针对PacBio HiFi Reads优化了组装流程),通过均聚物压缩,基于overlap的纠错和false overlap过滤来充分利用HiFi reads的全部潜能
- 使用30X PacBio HiFi Reads通过HiCanu将人CHM13细胞系的基因组组装的ContigN50提升至77M,单碱基准确性超过99.999%(Q50),在组装的准确性和连续性这两个方面,PacBio HiFi Reads的组装结果都超过了最新的高覆盖度超长牛津纳米孔测序的结果。
HiFiasm
Pub Date : 2020-08-03
可识别单倍型的纠错
Hifiasm会将所有的hifi reads读取到内存中进行all-vs-all比对并进行纠错。基于reads间的overlap信息,如果read上有一个碱基与其他碱基不同,并有至少3条reads支持,则认为它是SNP并保留,否则认为是错误并进行纠正。
组装图的构建
在校正之后,大多数错误被去除,同时杂合变异信息被保留。基于这些信息,Hifiasm构建了以reads为顶点、重叠区为边的定相string-graph。
组装序列的生成
如果没有其他数据,Hifiasm在输出序列时会任意选择每个气泡的一侧输出类似Falcon unzip和HiCanu的主要组装结果(primary contigs)。如果同时有父母本的测序数据,Hifiasm可以通过亲本特有的kmer在图上识别出来自父母本的序列,从而得到两套单倍体基因组。
当然HiFiasm文章中也提到了:
- 与其他基于图形的汇编程序不同,HiFiasm致力于保持所有单倍型的连续性。
- HiCanu只试图保持一个亲本单倍型的连续性,并且经常破坏另一个单倍型的连续性,当分离亲本单倍型时,这些突变点将导致单倍型分解的碎片—HiCanu没有充分利用HiFi Reads
- Hifiasm针对HiFi特点而开发,在hifi数据的组装表现上较同类软件更为突出,在多个基因组上表现出了更高的准确性和组装的连续性。
总结
No genome too large for HiFi reads.
参考文献
[1] Chen H , Zeng Y , Yang Y , et al. Allele-aware chromosome-level genome assembly and efficient transgene-free genome editing for the autotetraploid cultivated alfalfa[J]. Nature Communications, 2020, 11(1).
[2] Sequencing technologies — the next generation.[J]. Nature Reviews Genetics, 2010.
Korlach J , Bjornson K P , Chaudhuri B P , et al. Real-Time DNA Sequencing from Single Polymerase Molecules[J]. Methods in enzymology, 2010, 472:431-455.
[3] Cheng H, Concepcion G T, Feng X, et al. Haplotype-resolved de novo assembly with phased assembly graphs. arXiv 2020[J]. arXiv preprint arXiv:2008.01237, 2008.
[4] Nurk S, Walenz B P, Rhie A, et al. HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads[J]. BioRxiv, 2020.
[5] Wenger A M, Peluso P, Rowell W J, et al. Highly-accurate long-read sequencing improves variant detection and assembly of a human genome. bioRxiv[J]. Preprint]. doi, 2019, 10: 519025.
[6] Lang D, Zhang S, Ren P, et al. Comparison of the two up-to-date sequencing technologies for genome assembly: HiFi reads of Pacbio Sequel II system and ultralong reads of Oxford Nanopore[J]. bioRxiv, 2020.
[7] Wenger A M, Peluso P, Rowell W J, et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome[J]. Nature biotechnology, 2019, 37(10): 1155-1162.
[8] Sun X, Jiao C, Schwaninger H, et al. Phased diploid genome assemblies and pan-genomes provide insights into the genetic history of apple domestication[J]. Nature genetics, 2020, 52(12): 1423-1432.
{% btn ‘https://lixingze.xyz’,点击返回主页,far fa-hand-point-right,purple larger %}
标签:组装,测序,基因组,reads,HiFi,genome 来源: https://blog.csdn.net/m0_49960764/article/details/113481036